


一、法規制
「醫薬品醫療機器等の品質、有効性及び安全性の確保に関する法律」(略稱:「薬機法」)に基づき、日本の醫薬品と醫療機器の管理はこの法律に従います。「薬機法」は元の「薬事法」から変更されたもので、その具體的な実施規定には「薬機法施行令」、「QMS省令」およびその他の関連公告と通知が含まれます。
二、主管官庁
日本の醫薬品と醫療機器の管理は、主に厚生労働省(MHLW)と獨立行政法人醫薬品醫療機器総合機構(PMDA)の2つの主管官庁が擔當しています。
三、醫療機器分類
(1)一般醫療機器(ClassⅠ):厚生労働大臣が「薬事?食品衛生審議會」の意見を聴取した上で指定し、副作用や機能障害が発生した場合でも、人の生命や健康への影響リスクが比較的低い醫療機器です。
(2)管理醫療機器(ClassⅡ):同様に厚生労働大臣が「薬事?食品衛生審議會」の意見を聴取した上で指定し、副作用や機能障害が発生した場合、人の生命や健康に影響を與える可能性があるため、適切な管理が必要な醫療機器です。
(3)高度管理醫療機器(ClassⅢ、ClassⅣ):副作用や機能障害が発生した場合、人の生命や健康に重大な影響を與えるため、厳格な管理が必要な醫療機器です。

四、醫療機器認証モデルと登録審査プロセス
外國製造業者が日本に機器を輸出する場合、厚生労働省に登録申請を行う必要があります。この手続きは外國製造業者登録(FMR)と呼ばれ、PMD Act & 第169號令IS0 13485日本の醫療機器登録審査プロセスに従います。
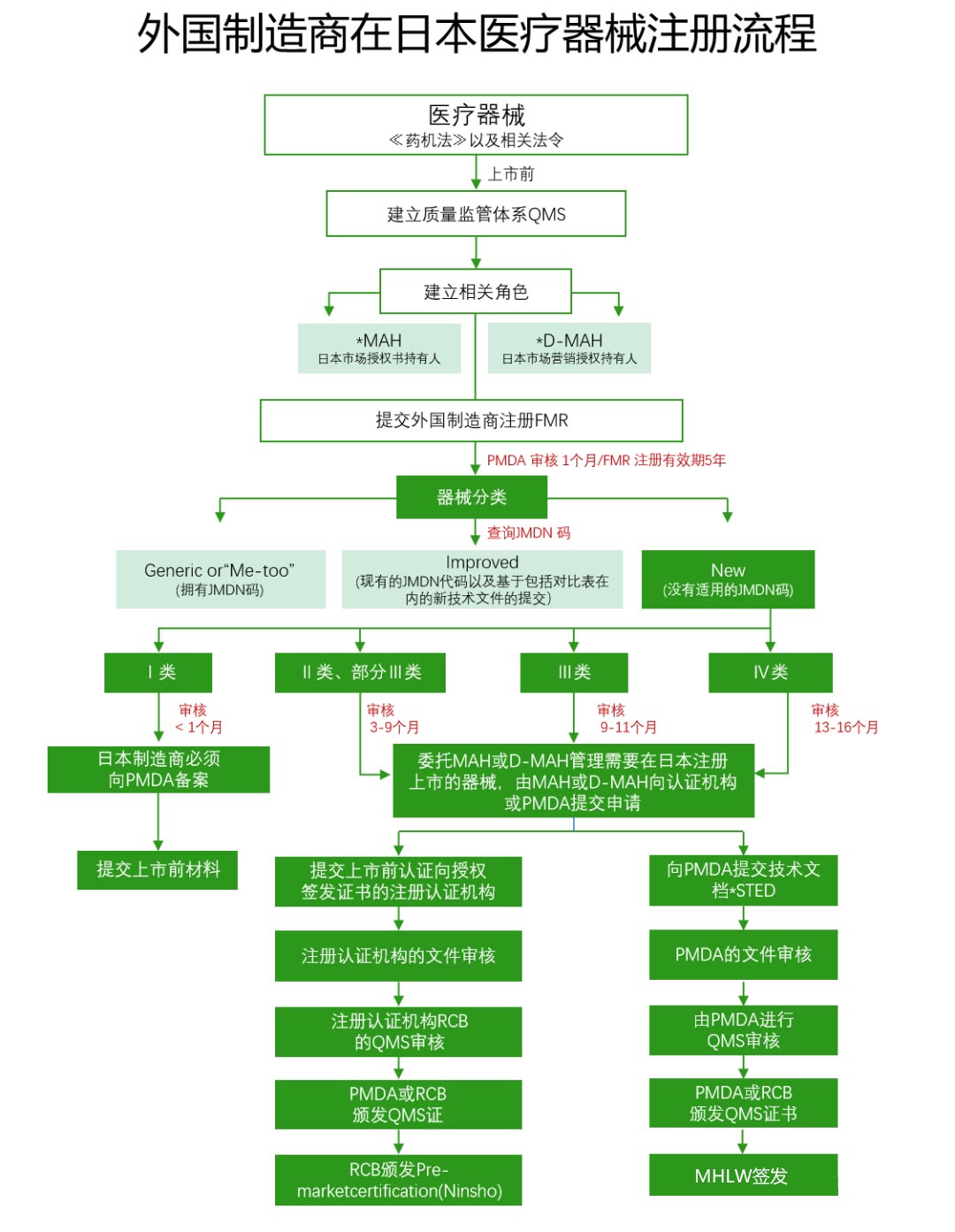
五、重點事項
(1)市場許認可保有者(MAH)/市場販売許認可保有者(D-MAH):外國製造業者は、日本で醫療機器を販売するための最初の條件として、日本で市場許認可保有者を指定する必要があります。
(2)STEDサマリー(登録資料):製品仕様、安定性と有効期限データ、性能試験データ、リスク分析、臨床データなどを含む必要があります。
(3)品質管理システム(QMS)監査:醫薬品醫療機器総合機構(PMDA)または登録認証機関(RCB)によって実施され、製造販売業者、醫療機器の設計、製造など、関連するすべての場所が含まれます。
(4)複數規制要件:日本市場に輸入される醫療機器は、電気用品安全法、電波法などの他の規制にも適合する必要があります。
 カスタマーサービスWeChatをフォローしてください
カスタマーサービスWeChatをフォローしてください